Early-Stage Medical Device Development Guide for Startups
A comprehensive regulatory framework for the early phases of medical device product development. Master the first phase of your development journey with expert guidance on user research, requirements development, and risk management.
Early-Stage Medical Device Development Guide for Startups
A Comprehensive Regulatory Framework for Initial Product Development
Version 1.0 | Based on FDA Guidance and Recognized Standards
Introduction and Scope
Purpose of This Guide
This guide provides startups with a structured, regulatory-compliant approach to early-stage medical device development. It focuses specifically on the critical transition period from concept exploration to formal design controls, addressing the activities necessary to establish indications for use, user personas, user needs, product requirements, and initial hazard analysis.
Unlike comprehensive design control or risk management guides, this resource targets the specific challenges startups face in the earliest phases of development—when resources are limited, regulatory requirements seem overwhelming, and critical decisions must be made that will impact the entire development trajectory.
What This Guide Covers
This guide addresses the following early-stage development activities:
- Pre-development research and feasibility - Activities that occur before formal design controls apply
- Intended use and indications for use development - Creating precise regulatory positioning statements
- User research and characterization - Understanding who will use your device and in what context
- User needs identification - Documenting what users need to accomplish
- Design input development - Translating user needs into engineering requirements
- Preliminary hazard analysis - Early identification of potential harms
- Initial formative evaluations - Early testing to inform design decisions
- Documentation framework establishment - Setting up traceability and control systems
What This Guide Does NOT Cover
To maintain focus and practical utility, this guide intentionally excludes:
- Complete design control implementation through design transfer
- Full risk management system implementation
- Comprehensive validation and verification protocols
- Manufacturing process development
- Clinical trial design and execution
- Regulatory submission preparation
- Post-market surveillance systems
Regulatory Foundation
This guide is built upon the following FDA guidance documents and recognized standards:
- FDA Design Control Guidance for Medical Device Manufacturers (March 1997)
- FDA Applying Human Factors and Usability Engineering to Medical Devices (February 2016)
- FDA General/Specific Intended Use Guidance (November 1998)
- ISO 14971:2019 - Medical devices - Application of risk management
- IEC 62366-1:2015 - Medical devices - Application of usability engineering
- ISO 13485:2016 - Medical devices - Quality management systems
- ANSI/AAMI HE75:2009/(R)2018 - Human factors engineering - Design of medical devices
- AAMI TIR59:2017 - Integrating human factors into design controls
How to Use This Guide
This guide follows a phase-based approach that mirrors the natural progression of medical device development. Each phase includes:
- Objectives - What you need to accomplish
- Activities - Specific tasks to complete
- Deliverables - Documents and artifacts to produce
- Decision Points - Gates where go/no-go decisions are made
- Templates - Structured formats for documentation
- Examples - Illustrative cases from various device types
Start with Phase 0 if you're still exploring feasibility. If you've already committed to product development, begin with Phase 1. Use the templates provided as starting points, adapting them to your specific device and organizational needs.
Early-Stage Development Process Flow
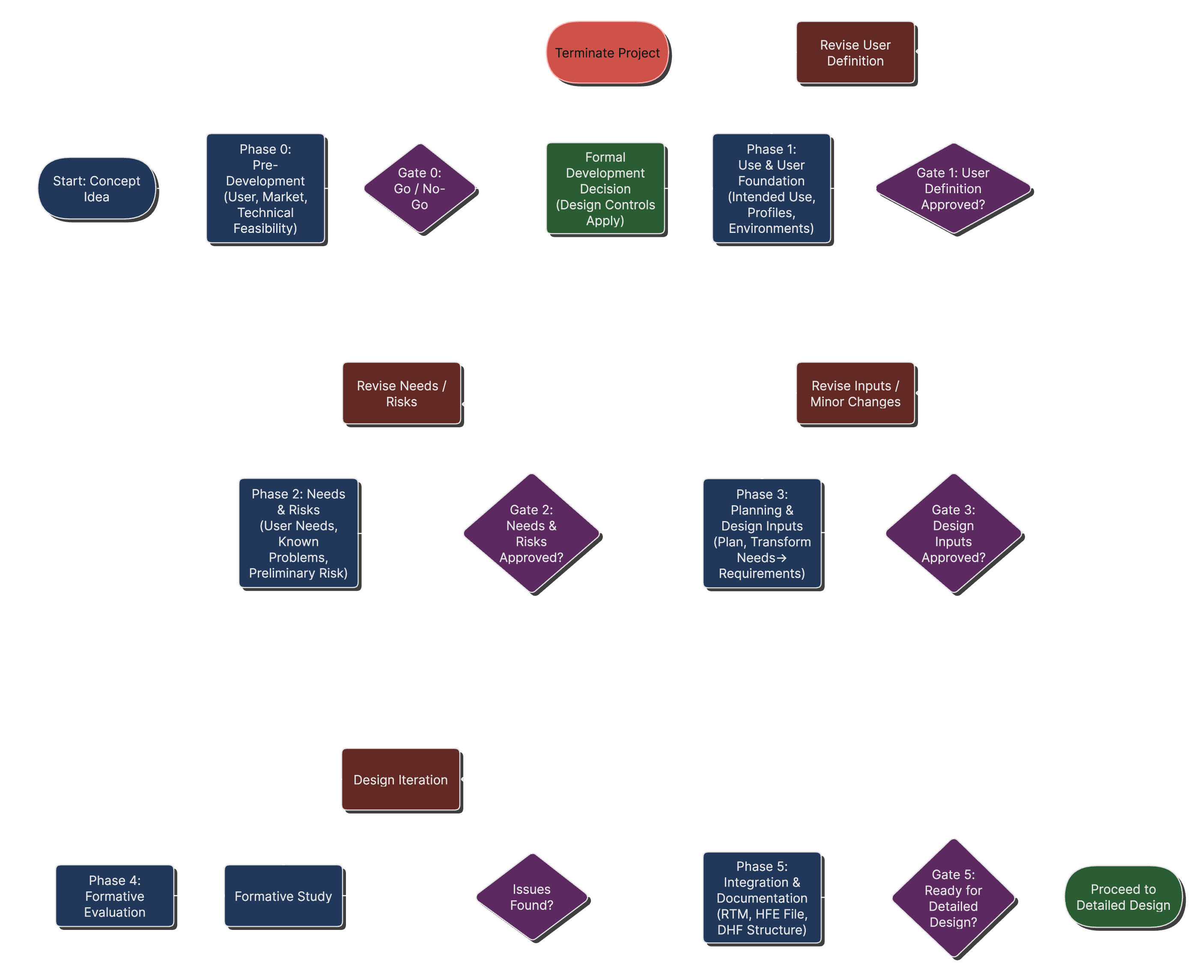
Early stage process flow with decision points on how to proceed.
Critical Success Factors
Before beginning your development journey, understand these critical success factors:
- Documentation Discipline - Even in early stages, maintain organized records
- User Focus - Ground all decisions in actual user needs, not assumed requirements
- Regulatory Strategy - Consider regulatory pathway implications from the start
- Risk Thinking - Integrate safety considerations into every decision
- Traceability - Establish clear links between needs, requirements, and controls
- Iterative Development - Plan for multiple design-test-refine cycles
Understanding the Regulatory Landscape
When Do Design Controls Apply?
One of the most critical decisions in early medical device development is determining when formal design controls must be implemented. The FDA Design Control Guidance provides clear direction on this transition point, but it's often misunderstood by startups.
Feasibility vs. Product Development
The FDA explicitly states that design controls do not apply to concept and feasibility activities. According to the Design Control Guidance (March 1997, page 14):
"This regulation does not apply to the research phase. Instead, the regulation requires companies that engage in the design and development of medical devices to establish procedures to control the design process."
This distinction is crucial for startups because it means you can explore concepts, build prototypes, and conduct user research without the full burden of design control documentation. However, once you make the decision to develop a specific device for commercialization, design controls become mandatory.
The Transition Trigger
The transition from feasibility to formal development occurs when management makes a conscious decision to develop a device for commercial distribution. This decision should be documented and typically includes:
- Market Commitment - Decision to pursue a specific market opportunity
- Resource Allocation - Assignment of dedicated personnel and budget
- Development Timeline - Establishment of project milestones
- Regulatory Strategy - Preliminary determination of classification and pathway
Design Control Trigger Points
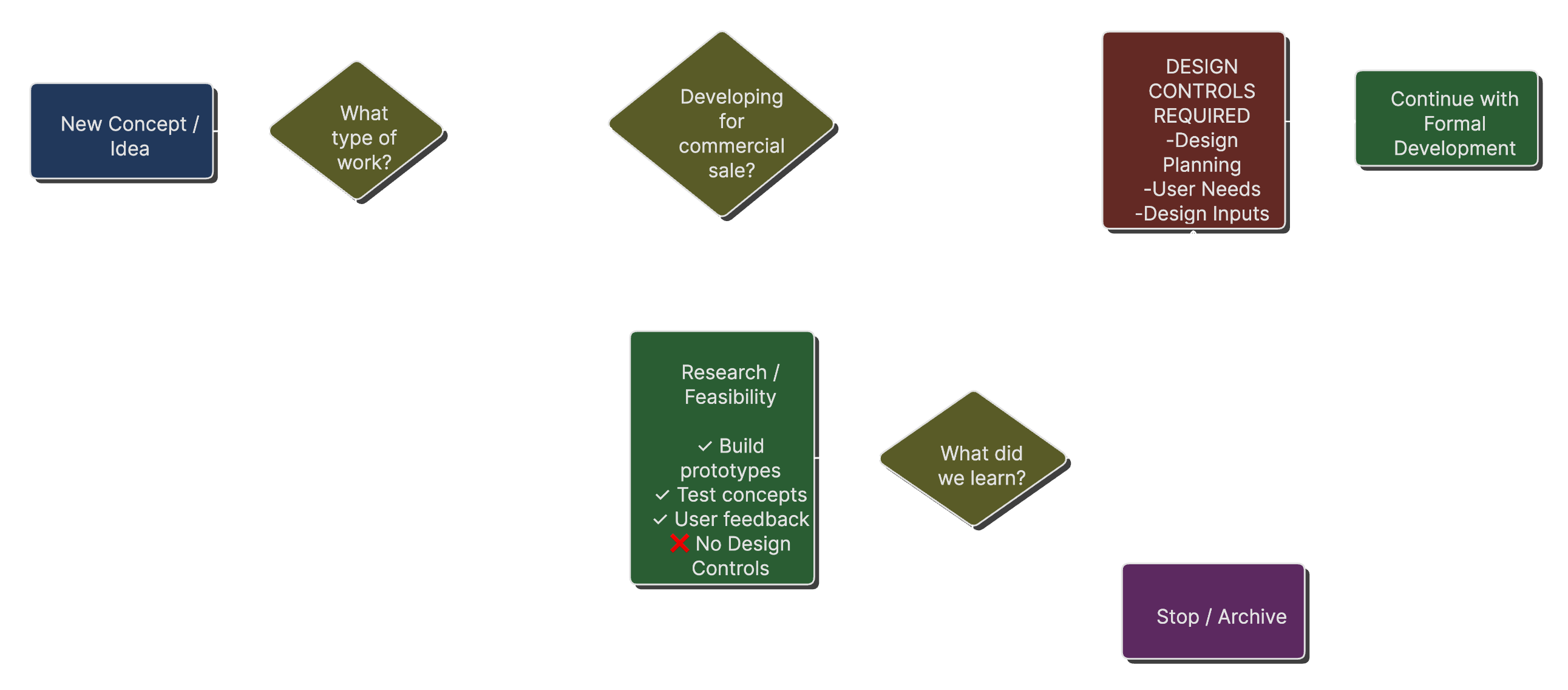
A decision tree showing when design controls apply versus when they don't, based on FDA guidance about feasibility versus product development.
Practical Implications for Startups
Understanding this transition point allows startups to:
- Conduct exploratory research without excessive documentation
- Build proof-of-concept prototypes for investor demonstrations
- Perform initial user studies to validate market need
- Iterate rapidly on early concepts
However, once you transition to formal development, retrospectively documenting design control activities is difficult and risky. Plan for this transition and ensure you have systems in place before crossing the threshold.
The User-Centered Design Imperative
Modern medical device development requires a user-centered approach from the earliest stages. This isn't just good practice; it's a regulatory expectation. The FDA's Human Factors Guidance (February 2016) states:
"FDA expects manufacturers to begin their human factors engineering process during the early stages of design and development."
This expectation is reinforced by IEC 62366-1:2015, which requires manufacturers to establish a use specification before beginning detailed design work. The use specification must include:
- Intended medical indication
- Intended patient population
- Intended user profile
- Use environment characteristics
- Operating principle
User Needs to Design Input Transformation
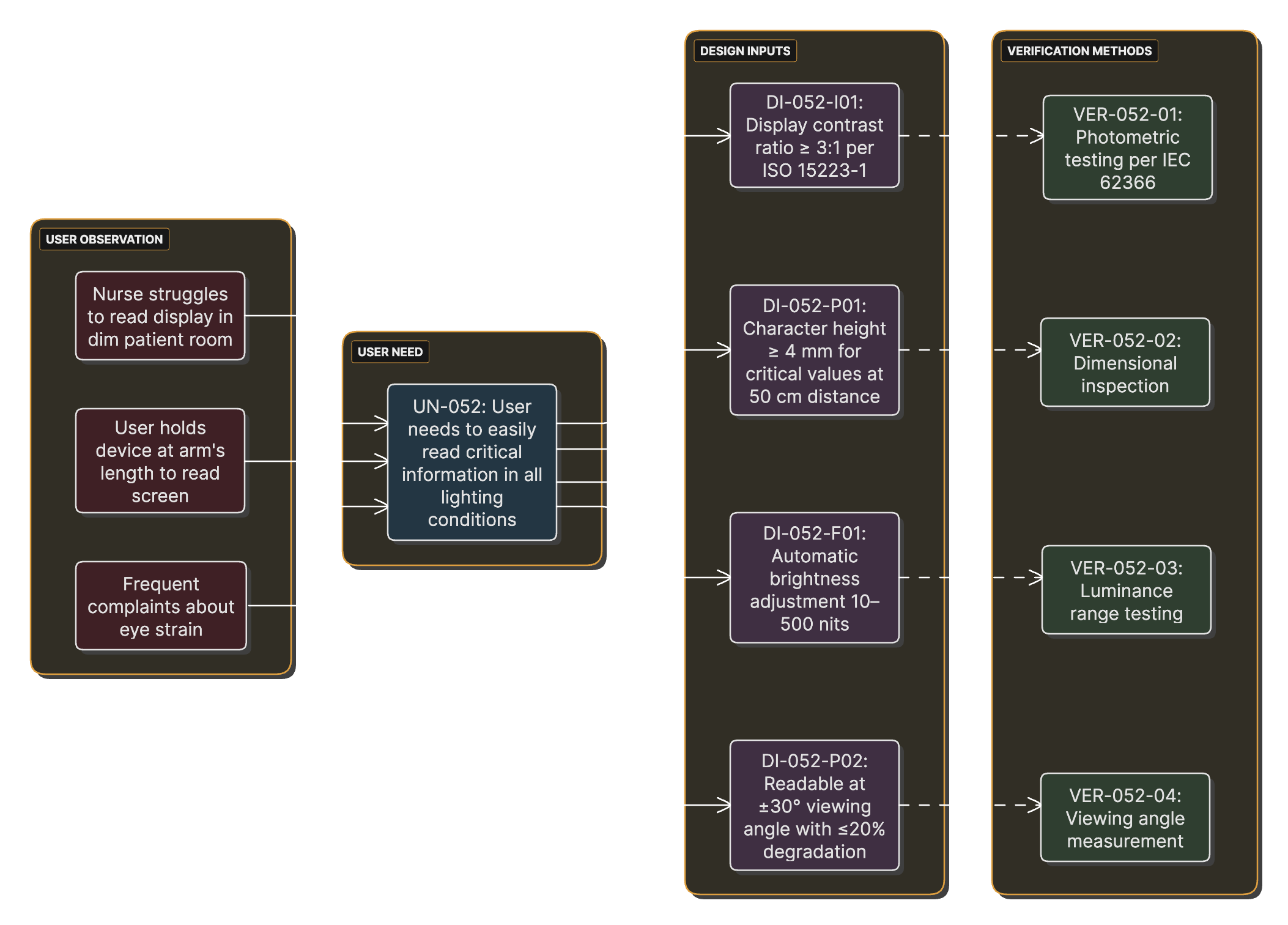
Over the course of a project, user observations should be documented as user needs that lead to design inputs that must be verified.
Why Early User Focus Matters
Starting with users in mind provides several advantages:
- Reduced Development Risk - Early identification of use-related hazards
- Lower Change Costs - Design modifications are less expensive before detailed development
- Better Market Fit - Products aligned with actual user needs
- Regulatory Efficiency - Fewer surprises during submission review
- Competitive Advantage - Superior usability can differentiate products
Risk Management from Day One
ISO 14971:2019 establishes risk management as a continuous process throughout the device lifecycle. For early-stage development, this means conducting preliminary hazard analysis (PHA) even before detailed design begins.
The PHA serves several purposes:
- Hazard Identification - Cataloging potential sources of harm
- Design Input Generation - Risk controls become design requirements
- Resource Planning - Understanding testing and validation needs
- Regulatory Strategy - Assessing classification and pathway implications
The PHA Advantage for Startups
Preliminary hazard analysis is particularly valuable for startups because:
- It can be performed with limited design detail
- It helps identify "showstopper" risks early
- It demonstrates regulatory sophistication to investors
- It guides technology selection and architecture decisions
- It prevents costly late-stage design changes
Phase 0: Pre-Development Foundation
Overview and Objectives
Phase 0 encompasses all activities that occur before the formal decision to develop a product for commercialization. This phase is characterized by exploration, learning, and validation of fundamental assumptions about market need and technical feasibility.
Primary Objectives:
- Validate market need and user interest
- Explore technical feasibility
- Understand the use environment and workflow
- Identify key stakeholders and their needs
- Build knowledge base for informed go/no-go decision
Regulatory Status: Activities in this phase are NOT subject to design controls per FDA guidance. However, maintaining good documentation practices will facilitate the transition to formal development.
User Research Activities
Contextual Inquiry
Contextual inquiry involves observing users in their actual work environment as they perform relevant tasks. This method, referenced in FDA Human Factors Guidance Section 6.4.1, provides insights that cannot be obtained through interviews or surveys alone.
Example Observation Recording Sheet
## Contextual Inquiry Observation Sheet **Date:** ___________ **Observer:** ___________ **Facility:** ___________ **User Role:** ___________ ### Task Observed: [Specific task name] **Start Time:** _____ **End Time:** _____ **Environment Characteristics:** - Lighting: [ ] Bright [ ] Moderate [ ] Dim - Noise Level: [ ] Quiet [ ] Moderate [ ] Loud - Interruptions: [ ] None [ ] Few [ ] Many - Space: [ ] Spacious [ ] Adequate [ ] Cramped **Task Flow:** 1. Step performed: _____________________ - Time taken: _____ - Difficulties observed: _____ - Workarounds used: _____ **Critical Observations:** - Safety concerns: _____________________ - Efficiency issues: ___________________ - User frustrations: ___________________
User Interviews and Focus Groups
While contextual inquiry provides behavioral insights, interviews and focus groups reveal attitudes, preferences, and expectations. FDA Human Factors Guidance Section 6.4.2 identifies these as valuable early-stage research methods.
Technical Feasibility Assessment
While user research validates market need, technical feasibility confirms the ability to meet that need within practical constraints.
Technical Feasibility Assessment Matrix
## Technical Feasibility Assessment ### Core Technical Requirements | Requirement | Feasible? | Risk Level | Notes | |------------|-----------|------------|--------| | Measurement accuracy ±2% | Yes | Low | Demonstrated with breadboard | | 8-hour battery life | Maybe | Medium | Requires low-power design | | <$50 manufacturing cost | No | High | Current estimate $85 | | Waterproof to IPX7 | Yes | Medium | Requires sealing redesign | | 5-year shelf life | Unknown | High | Requires stability testing |
Decision Point: Transition to Formal Development
The transition from Phase 0 to Phase 1 represents a critical business decision with regulatory implications. This decision should be formal, documented, and based on clear criteria.
Go/No-Go Decision Criteria
- Market Validation: Confirmed user need through research (minimum 10 users)
- Technical Feasibility: Core technology demonstrated
- Regulatory Pathway: Device classification determined
- Resources: Development funding secured/available
- Risk Assessment: No unacceptable safety risks identified
Gate Review Decision Matrix
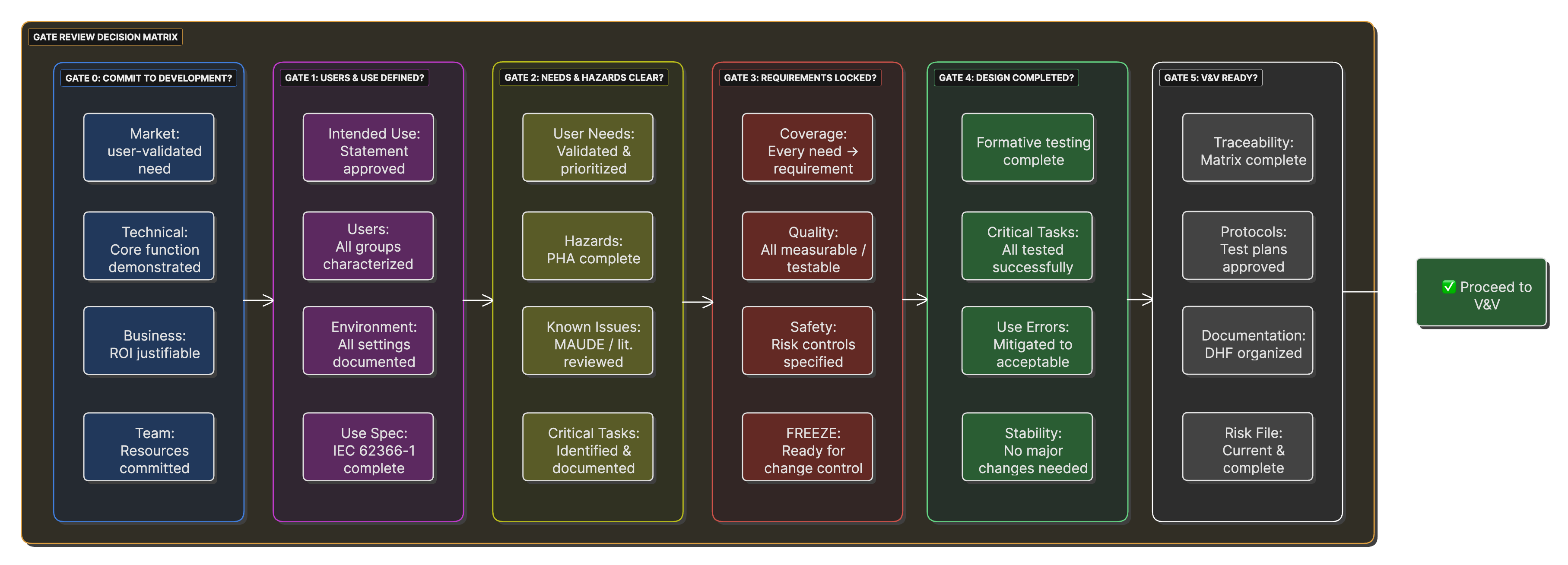
A matrix showing criteria for each gate review, with go/no-go decision factors and required documentation for passage.
Unlock the Complete Guide
Get instant access to the full Early-Stage Medical Device Development Guide including comprehensive phase guidance, templates, examples, and expert insights on risk management, regulatory compliance, and best practices.
- Complete Phase 1-5 development methodology
- 15+ Copy-paste templates for immediate use
- Worked examples from real FDA submissions
- Risk management frameworks & checklists
- Design controls and traceability guidance
- Common pitfalls and how to avoid them
- Complete regulatory reference list
Enter your details to unlock the complete 18,000-word guide immediately. No spam, unsubscribe anytime.
Phase 1: Establishing Use and User Foundation
Overview and Transition
Phase 1 marks the beginning of formal product development. With the decision to proceed, you must now establish the foundational elements that will guide all subsequent development activities. This phase focuses on precisely defining WHO will use your device, WHERE they will use it, and WHAT medical purpose it serves.
Critical Transition Note
From this point forward, design control requirements apply. All outputs from Phase 1 become part of your Design History File (DHF) and must be properly controlled, reviewed, and approved.
Developing the Intended Use Statement
The intended use statement is arguably the most important regulatory document you'll create. It determines your device classification, regulatory pathway, and the scope of FDA review. The FDA's General/Specific Intended Use Guidance provides a framework for crafting these statements.
Understanding Levels of Specificity
The FDA guidance describes increasing levels of specificity for intended use statements:
Template for Intended Use Statement
## Intended Use Statement ### Device Name: [Your Device Name] ### Intended Use: The [Device Name] is intended to [primary function/measurement] in [target population] for [medical purpose] in [use environment]. ### Indications for Use: The [Device Name] is indicated for [specific medical condition or use case] in [specific patient population with relevant characteristics]. The device is intended for use by [user types] in [specific settings]. ### Key Constraints/Limitations: - Not intended for [specific exclusion] - Not for use in [excluded population] - Should not be used for [excluded purpose] ### Prescription/OTC Status: [ ] Prescription Use (Rx) [ ] Over-the-Counter (OTC)
User Characterization and Personas
Understanding your users in detail is essential for both design success and regulatory compliance. IEC 62366-1:2015 requires formal documentation of user characteristics that might impact safe use.
Stakeholder Map for Medical Device Development
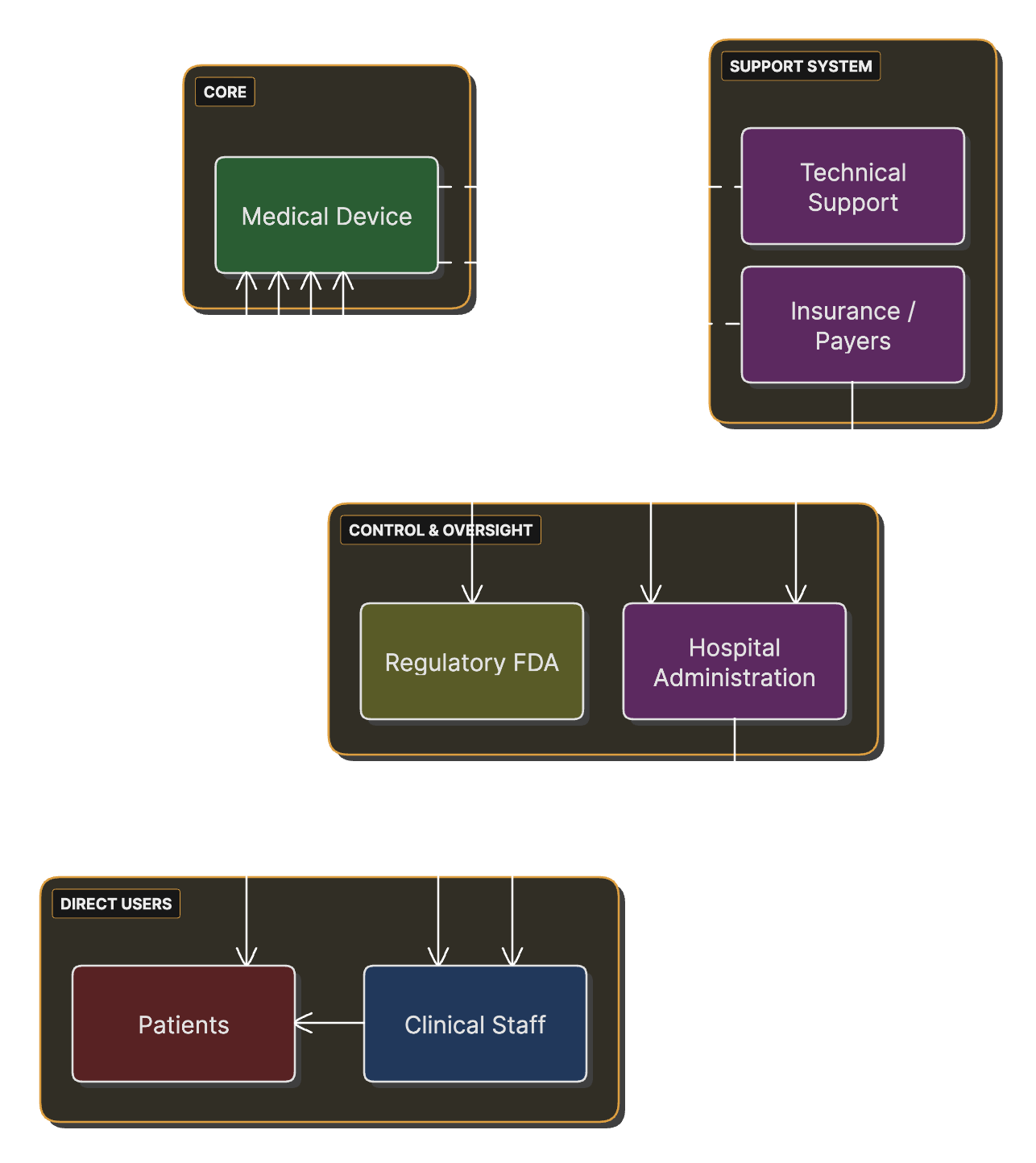
Stakeholders (users, patients, caregivers, facilities, regulators) and their relationships to the device.
Example Persona: "Nurse Nancy"
Background: Nancy is a 42-year-old registered nurse with 18 years of experience in medical-surgical units. She works 12-hour shifts, typically caring for 5-6 patients per shift.
Goals:
- Minimize time spent on routine tasks
- Reduce documentation burden
- Prevent medication errors
- Provide compassionate patient care
Key Design Implications:
- Minimize interaction steps
- Large, clear touch targets
- Glove-compatible interface
- Audible feedback in noisy environment
Use Environment Specification
The use environment significantly impacts device design and safety. FDA Human Factors Guidance Section 5.2 requires comprehensive environment characterization.
Creating the Use Specification
The Use Specification, required by IEC 62366-1:2015, synthesizes user and environment information into a formal document that drives human factors engineering activities.
Phase 2: User Needs and Initial Risk Identification
Overview
Phase 2 transforms user research insights into documented needs and identifies potential hazards associated with device use. This phase creates the foundation for all subsequent design decisions and establishes the safety framework that will guide development.
User Needs Elicitation and Documentation
User needs are high-level, qualitative statements that describe what users need to accomplish, independent of specific design solutions. FDA Design Control Guidance emphasizes that user needs are NOT design inputs—they must be translated into measurable requirements in Phase 3.
User Need Documentation Structure
## User Need Documentation ### Need Identifier: UN-XXX **Need Statement:** The [specific user type] needs to [accomplish something specific] [under what conditions] [to achieve what outcome]. **User Type:** [Primary User/Secondary User/Patient/Caregiver] **Source:** - [ ] Contextual inquiry observation - [ ] User interview - [ ] Focus group - [ ] Complaint/incident analysis **Priority:** - [ ] Critical - Essential for safe and effective use - [ ] High - Significant impact on user satisfaction - [ ] Medium - Improves user experience - [ ] Low - Nice to have **Acceptance Criteria:** [How will we know this need is met?]
Preliminary Hazard Analysis (PHA)
The PHA identifies potential hazards before detailed design begins, allowing risk mitigation to be built into the design from the start. ISO 14971:2019 Clause 5 provides the framework for this analysis.
Risk Management Integration Timeline
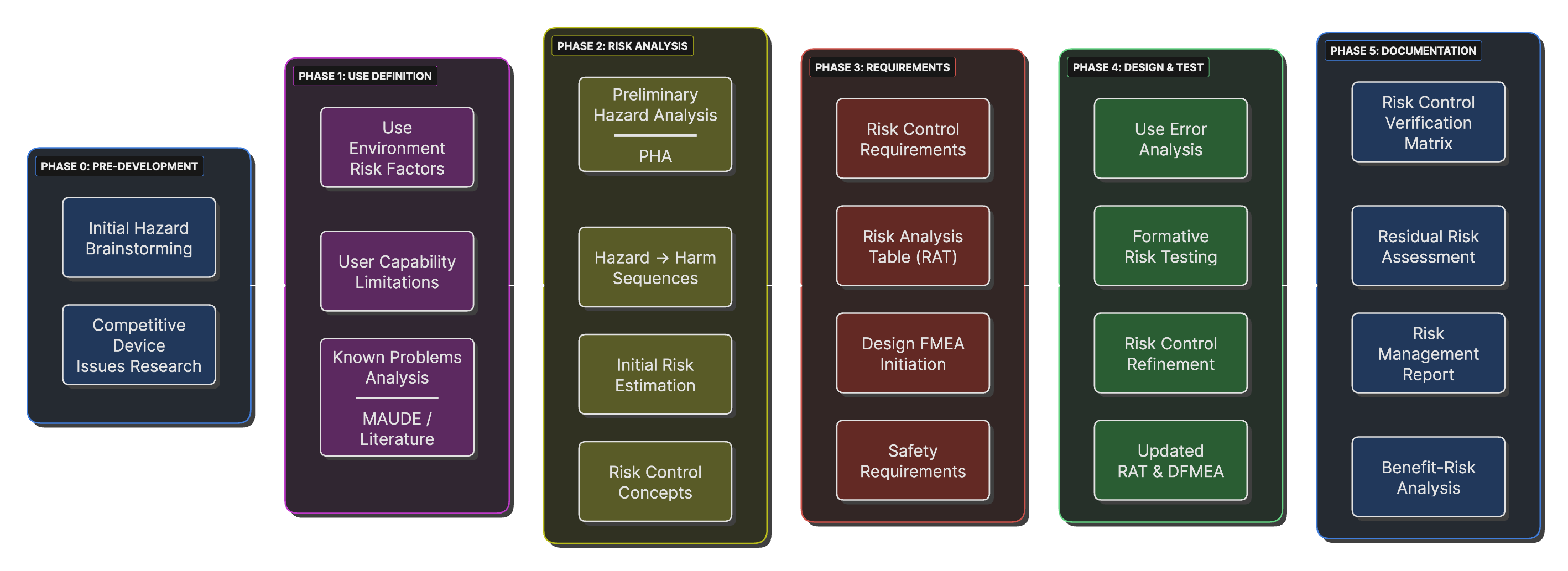
A timeline showing when different risk management activities occur relative to design control milestones, emphasizing early hazard identification.
Hazard Identification Checklist
## Hazard Identification Checklist ### Energy Hazards - [ ] Electrical energy (shock, burn) - [ ] Thermal energy (hot/cold surfaces) - [ ] Mechanical energy (moving parts, sharp edges) - [ ] Radiation (ionizing, non-ionizing) - [ ] Acoustic energy (loud sounds) ### Biological Hazards - [ ] Biocontamination (bacteria, viruses) - [ ] Biocompatibility (material toxicity) - [ ] Cross-contamination between patients ### Chemical Hazards - [ ] Toxic materials - [ ] Cleaning agent residues - [ ] Battery chemicals - [ ] Allergens ### Operational Hazards - [ ] Use error (foreseeable misuse) - [ ] Inadequate information - [ ] Confusion with other devices - [ ] Software failures
Known Problems Analysis
Research existing similar devices to identify documented problems and ensure your design addresses known issues.
Integration of Needs and Risks
User needs and risks must be considered together to ensure the design addresses both user requirements and safety concerns.
Phase 3: Design Planning and Input Development
Overview
Phase 3 transforms user needs and risk analyses into formal design requirements. This phase establishes the technical specifications that will guide detailed design and creates the framework for project management and control.
Design and Development Planning
Per FDA Design Control Guidance Section B and ISO 13485:2016 Section 7.3.2, a formal design plan must be established before beginning detailed design work.
Document Hierarchy and Traceability
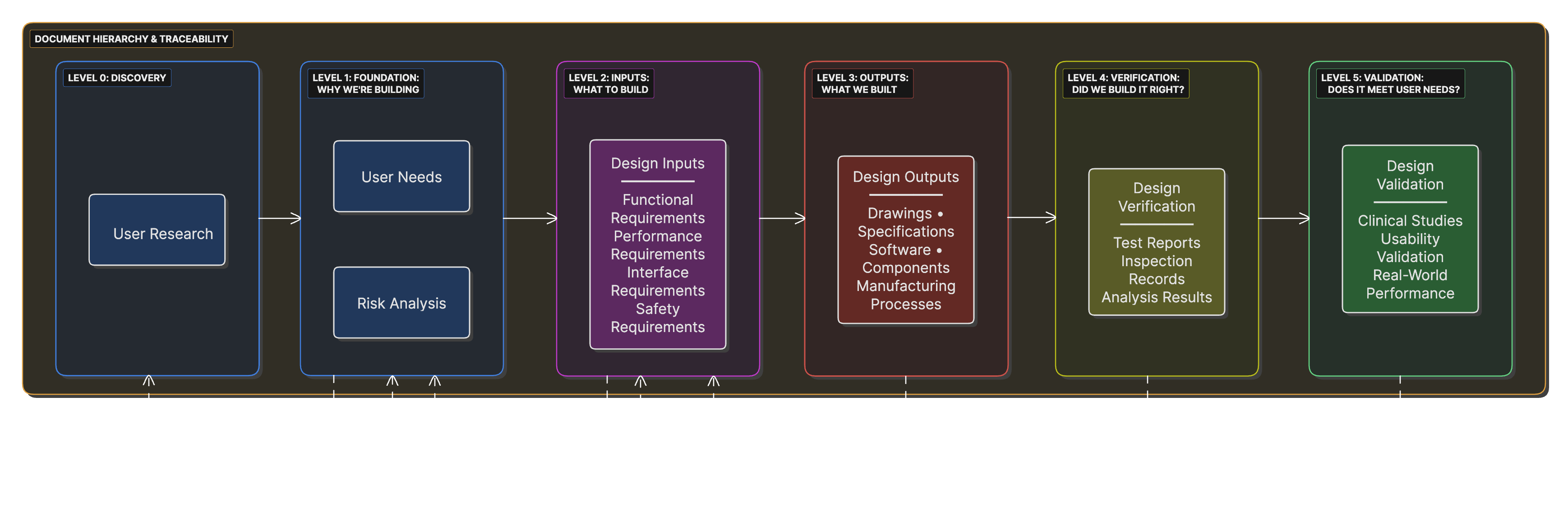
A pyramid structure showing the relationship between user needs, design inputs, design outputs, and verification/validation, with bidirectional traceability arrows.
Design Input Requirements Development
Design inputs are the technical specifications that the device must meet. FDA Design Control Guidance Section C provides detailed guidance on developing adequate design inputs.
User Need to Design Input Translation
## User Need to Design Input Translation ### User Need: UN-042 "The emergency room nurse needs to obtain patient vital signs within 60 seconds of patient arrival to enable rapid triage decisions." ### Analysis: **What must the device do?** (Functional requirement) - Measure blood pressure, pulse, temperature, SpO2 **How well must it perform?** (Performance requirement) - Complete all measurements within 60 seconds - Achieve specified accuracy for each parameter ### Resulting Design Inputs: #### DI-042-F01: Functional Requirement **Title:** Vital Signs Measurement Capability **Requirement:** The device shall measure systolic blood pressure, diastolic blood pressure, pulse rate, body temperature, and SpO2. **Verification Method:** Demonstration of each measurement capability **Accept Criteria:** All five parameters successfully measured
Design Input Categories and Examples
Design inputs typically fall into four main categories:
- Functional Requirements - What the device must do
- Performance Requirements - How well it must perform
- Interface Requirements - How it interacts with users and systems
- Safety Requirements - Safety features and characteristics
Design Input Review and Approval
Design inputs must be formally reviewed and approved before proceeding to detailed design. This review ensures requirements are complete, unambiguous, and verifiable.
Phase 4: Early Formative Evaluation and Risk Control Planning
Overview
Phase 4 focuses on evaluating early design concepts through formative studies and planning risk control measures. This iterative phase helps refine the design before committing to expensive detailed development.
Formative Evaluation Planning
Formative evaluations, as described in FDA Human Factors Guidance Section 6.4.3, are studies conducted during development to inform and improve the design.
Formative vs Summative Evaluation
Side-by-side comparison for usability engineering decisions.
| Category | Formative – Improve the Design | Summative – Validate the Design |
|---|---|---|
| WHEN | During developmentMultiple iterations |
End of developmentSingle study |
| PURPOSE | Find problems Inform design decisions |
Prove safety Validate effectiveness |
| PARTICIPANTS | 5–10 per roundCan reuse participants |
15+ per user groupRepresentative only |
| PROTOTYPE | Any fidelity Mockups → working models |
Production-equivalent Final design frozen |
| METHOD | Think-aloud Discussion encouraged Assistance allowed |
Realistic use No assistance Performance metrics |
| OUTCOME | List of improvements Design changes expected |
Pass / Fail FDA submission data |
Formative Evaluation Plan Template
## Formative Evaluation Plan ### Study Identifier: FE-001 **Study Title:** [Descriptive title] **Study Type:** [Cognitive walkthrough/Simulated use/Expert review] ### Objectives Primary Objective: [What specific aspect of design is being evaluated] ### Design Elements Under Evaluation - Element 1: [Specific interface component or feature] - Questions to answer: _______ - Success criteria: _______ ### Methodology **Target N:** _______ **User Groups:** - Group 1: n=___, Characteristics: _______ **Task Scenarios:** 1. Scenario 1: [Normal use case] 2. Scenario 2: [Challenging case] ### Success Criteria **Overall Success:** - >80% task completion without assistance - No critical use errors observed - Positive user feedback on key elements
Risk Control Planning
Based on the PHA from Phase 2, develop specific risk control measures following ISO 14971 hierarchy.
Critical Task Identification Process
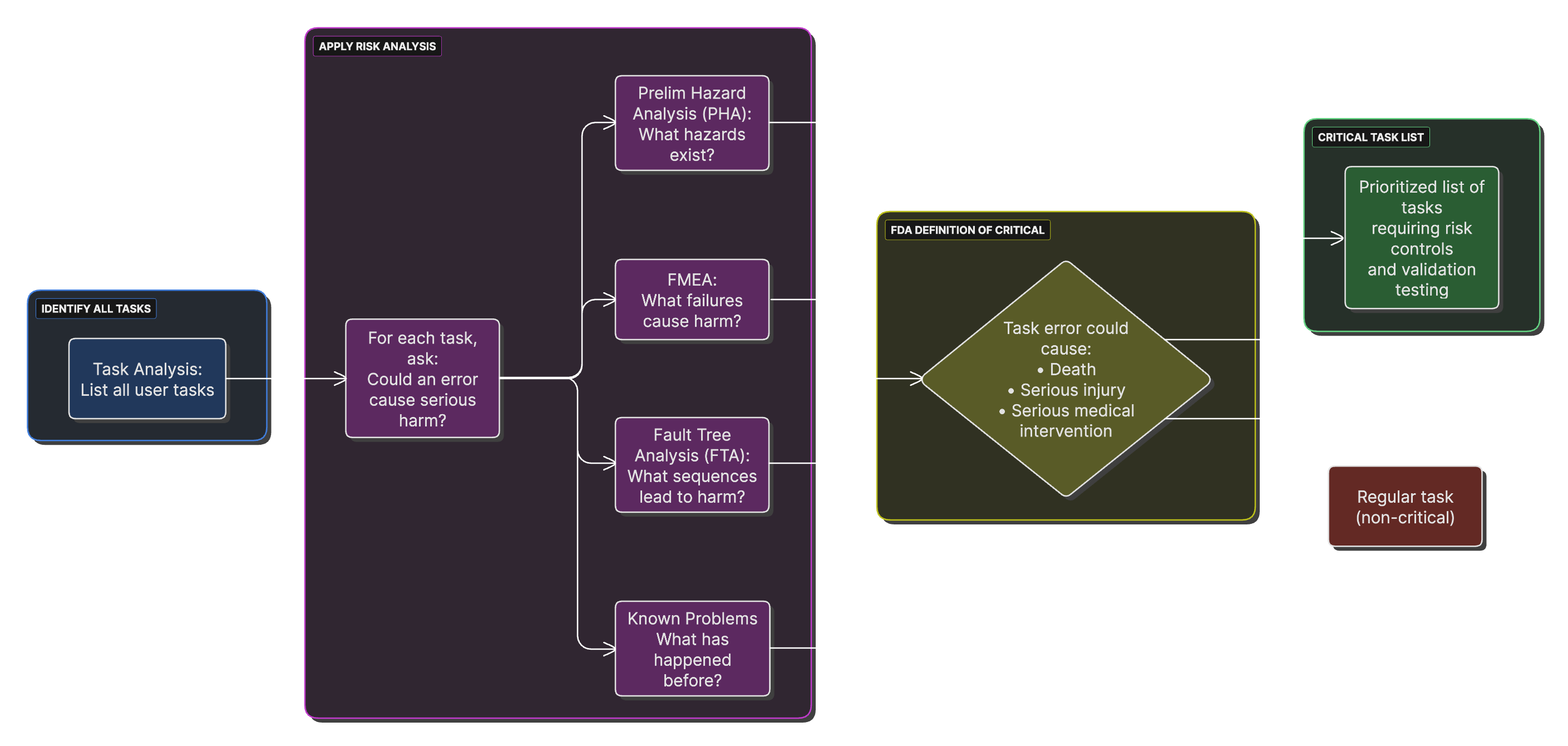
A process flow showing how to identify critical tasks through task analysis, FMEA, and fault tree analysis, converging into a prioritized critical task list.
Integration of Formative Results
Formative evaluation results must be systematically incorporated into the design.
Phase 5: Integration and Documentation
Overview
Phase 5 establishes the framework for comprehensive documentation and traceability that will carry through the remainder of development. This phase ensures all early-stage work is properly documented and linked.
Requirements Traceability Matrix Development
The Requirements Traceability Matrix (RTM) is the master document linking user needs through to validation.
Comprehensive RTM Template
## Requirements Traceability Matrix ### Forward Traceability (Needs → Validation) | User Need | Design Input | Design Output | Verification | Validation | Risk Control | |-----------|--------------|---------------|--------------|------------|--------------| | UN-001: Rapid results | DI-015-P01: Results <60sec | SW-SPEC-023: Algorithm | VER-015: Timing study | VAL-002: Clinical study | RCM-008: Timeout alert | | UN-002: Readable display | DI-022-I03: Contrast >3:1 | DWG-045: Display spec | VER-022: Testing | VAL-002: Clinical study | RCM-012: Brightness | ### Coverage Analysis #### User Needs Coverage: - Total User Needs: 45 - Addressed by Design Inputs: 45 - Coverage: 100% #### Risk Controls Coverage: - Total Identified Risks: 23 - Risks with Controls: 23 - Coverage: 100%
Human Factors Engineering File
The HFE File documents all human factors activities and serves as the primary submission document for FDA human factors review.
Design History File Organization
The Design History File (DHF) must be established early and maintained throughout development.
Use Environment Factors Web
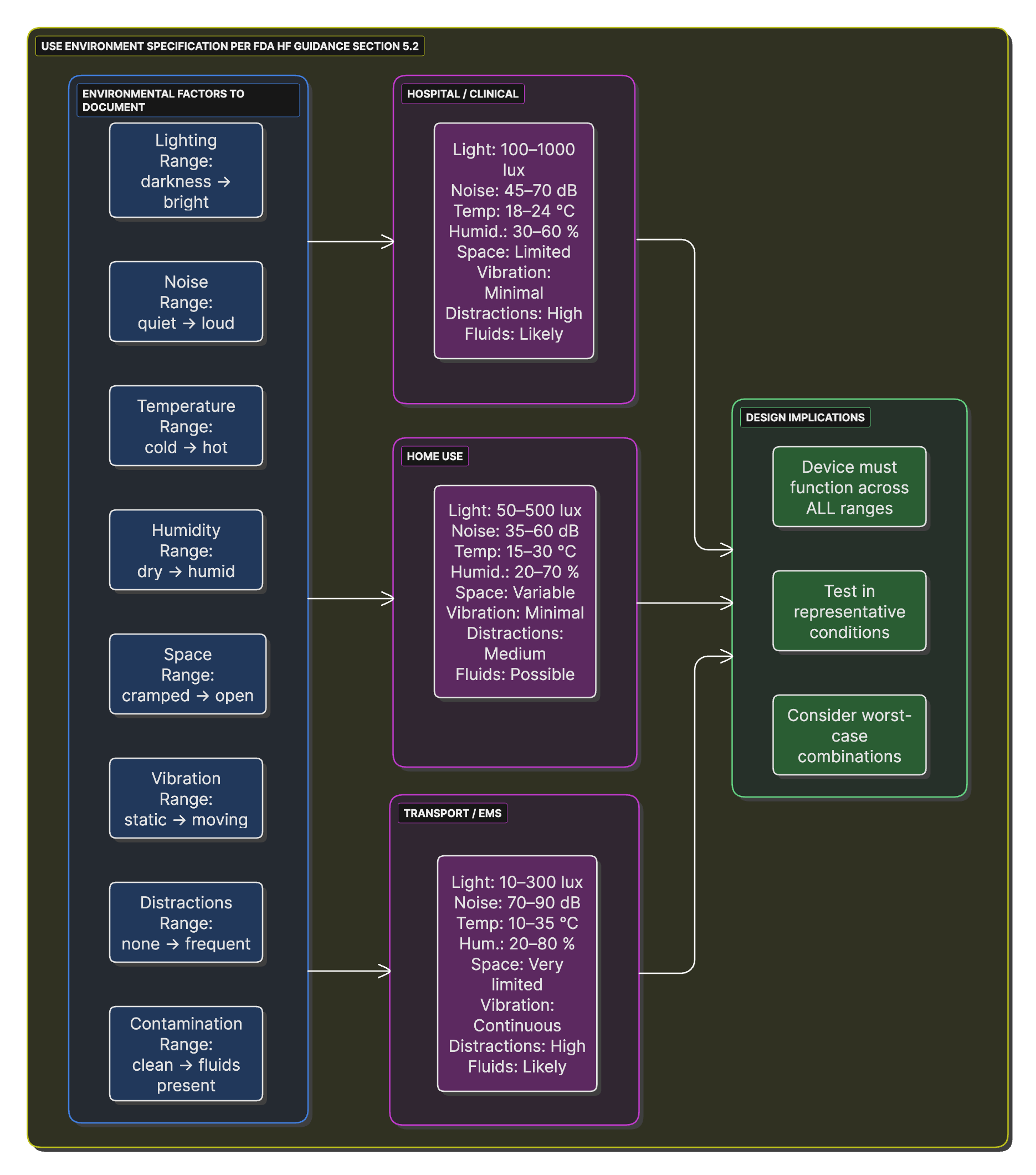
A spider diagram showing all environmental factors that must be considered (lighting, noise, temperature, etc.) with scales for different use environments.
Templates and Examples
Tip: Paste each template into your documentation system and fill it in. Keep IDs stable for traceability.
Template A — Intended Use vs. Indications for Use
# Intended Use vs. Indications for Use **Device working name:** **Regulatory pathway (anticipated):** 510(k) / De Novo / PMA / TBD ## Intended Use (single sentence) [Describe the general purpose/function of the device without specific patient population or settings.] ## Indications for Use (Form FDA 3881 style) [Condition/problem], for [intended patient population], used by [intended user] in [use environment(s)], to [purpose]. Key limitations/contraindications (if any): [list].
Template B — User Profile Documentation
## User Profile: [User Type Name] ### Role Definition **Primary Responsibilities:** - Responsibility 1: _____________ - Responsibility 2: _____________ **Frequency of Device Use:** - [ ] Multiple times daily - [ ] Daily - [ ] Weekly - [ ] Occasionally ### Demographics **Age Range:** _____________ **Gender Distribution:** _____________ ### Physical Characteristics **Vision:** - Typical acuity range: _____________ - Common conditions: _____________ **Dexterity/Strength:** - Fine motor skills: _____________ - Grip strength range: _____________ ### Cognitive Characteristics **Education Level:** - Minimum: _____________ - Typical: _____________ **Technical Literacy:** - Experience with similar devices: _____________
Template C — Preliminary Hazard Analysis Worksheet
## Preliminary Hazard Analysis Worksheet ### Analysis Information **Product:** [Device name and version] **Analysis Date:** _______ **Analysts:** [List team members] ### Hazard Analysis Table | ID | Hazard | Cause(s) | Hazardous Situation | Harm | Severity | Probability | Risk Level | |----|--------|----------|-------------------|------|----------|-------------|------------| | PH-001 | Electrical shock | Insulation failure | User contacts live parts | Burns, cardiac arrest | Critical | Remote | HIGH | | PH-002 | Sharp edge | Manufacturing defect | User cuts hand | Laceration | Minor | Occasional | LOW | ### Severity Categories **Catastrophic (5):** Death or permanent life-threatening injury **Critical (4):** Permanent impairment or life-threatening injury **Serious (3):** Injury requiring medical intervention **Minor (2):** Temporary injury or impairment **Negligible (1):** No injury or slight injury
Template D — Design Input Requirement
## Design Input Requirement ### Requirement ID: DI-XXX-YZZ ### Basic Information **Title:** [Concise descriptive title] **Category:** [Functional/Performance/Interface/Safety] **Priority:** [Critical/High/Medium/Low] **Status:** [Draft/Under Review/Approved] ### Requirement Statement The [device/system/component] shall [single specific requirement stated in clear, verifiable terms]. **Quantitative Specification** (if applicable): - Parameter: [What is being measured] - Value: [Specific number with tolerance] - Units: [Measurement units] - Conditions: [Test conditions, environment] ### Rationale **Source:** [UN-XXX user need or PH-XXX risk reference] **Justification:** [Why this specific requirement level] ### Verification **Method:** Test/Inspection/Analysis/Demonstration **Protocol Reference:** VER-XXX **Acceptance Criteria:** [Specific pass/fail criteria]
Template E — User Interview Guide
## Semi-Structured User Interview Guide ### Introduction Script "Thank you for participating. We're developing a new medical device and want to understand your needs and current practices. There are no right or wrong answers. This will take about 45 minutes." ### Section 1: Background (10 minutes) 1. Can you describe your current role and responsibilities? 2. What types of [relevant procedures] do you perform? 3. What is your experience with [current devices/methods]? ### Section 2: Current Workflow (15 minutes) 4. Walk me through a typical [procedure] from start to finish. 5. What works well in your current process? 6. What challenges or frustrations do you experience? ### Section 3: Specific Scenarios (10 minutes) 7. How do you handle [challenging scenario 1]? 8. Tell me about a time when [error/problem occurred]. ### Section 4: Ideal Solution (10 minutes) 9. If you could design the perfect [device], what would it do? 10. What should definitely be avoided in a new design?
Common Pitfalls and Best Practices
Common Pitfalls in Early Development
Pitfall 1: Starting Design Before Understanding Users
The Problem
Many startups jump directly to technical development based on assumptions about user needs, leading to products that technically work but fail in real use.
Warning Signs
- No direct user observation conducted
- Requirements based solely on competitive products
- Engineering team making clinical decisions
- "We know what users need" attitude
Best Practice
Spend at least 20% of early development time on user research. The FDA Human Factors Guidance explicitly expects user understanding to precede design.
Pitfall 2: Confusing User Needs with Design Solutions
Wrong Approach
"User needs a touchscreen interface" (This is a solution)
Right Approach
"User needs to interact with device while wearing gloves" (This is a need)
Best Practice
Always ask "why" when documenting needs. The real need is usually found after 3-5 "why" iterations.
Pitfall 3: Inadequate Risk Analysis Scope
The Problem
Focusing only on device failure modes while ignoring use errors and environmental factors.
Incomplete Risk Analysis
- Only considers component failures
- Ignores user interaction risks
- Misses environmental hazards
- Overlooks maintenance/service risks
Comprehensive Approach
- Device failures AND use errors
- Normal use AND foreseeable misuse
- Direct harm AND indirect consequences
- Patient AND user AND bystander risks
Pitfall 4: Creating Unverifiable Requirements
Examples of Poor Requirements
- "The device shall be user-friendly" (How do you measure this?)
- "The display shall be easily readable" (Under what conditions?)
- "The device shall operate reliably" (What's the metric?)
Examples of Good Requirements
- "The device shall have all controls accessible within a 40cm reach envelope"
- "The display shall have contrast ratio ≥3:1 at 500 lux ambient lighting"
- "The device shall operate for 10,000 cycles with <2% failure rate"
Pitfall 5: Losing Traceability
The Problem
Requirements evolve but links to original needs are lost, making it impossible to verify that user needs are met.
Prevention Strategies
- Establish numbering system from day one
- Document rationale for every requirement
- Track changes with reasons
- Regular traceability audits
- Single source of truth for requirements
Best Practices for Startup Success
Best Practice 1: Front-Load User Research
Investment Allocation
- Phase 0-1: 30% of resources on user research
- Phase 2-3: 20% on requirements development
- Phase 4+: 50% on design and implementation
ROI Evidence
FDA citations for human factors issues decreased 75% when companies conducted early user research (FDA CDRH data).
Best Practice 2: Iterate Early and Often
Iteration Strategy
- Paper prototypes: Daily iterations
- Mockups: Weekly iterations
- Working prototypes: Bi-weekly iterations
- Production design: Monthly reviews
Cost Multiplication Rule
Changes cost 10x more at each phase:
- Concept phase: $100
- Design phase: $1,000
- Verification phase: $10,000
- Post-market: $100,000
Best Practice 3: Document Decisions, Not Just Outcomes
Design Decision Record Template
## Design Decision Record **Decision ID:** DD-017 **Date:** 2024-03-20 **Decision:** Use rechargeable lithium-ion battery **Alternatives Considered:** 1. Disposable alkaline batteries 2. Rechargeable NiMH battery 3. Rechargeable lithium-ion battery 4. AC power only **Decision Rationale:** Selected Option 3 (Li-ion) because: - 8-hour runtime meets UN-032 (full shift operation) - Rechargeable reduces operating cost 90% - Energy density enables portable form factor - Safety risks manageable with protection circuits **Risk Mitigation:** - Implement battery management system - Use IEC 62133 compliant cells - Add thermal protection **Approvers:** Technical Lead: [Name] Quality: [Name] Regulatory: [Name]
Best Practice 4: Build Quality Into Design
Quality by Design Principles
- Error Prevention Over Error Detection
- Physical constraints preventing wrong connections
- Software interlocks preventing dangerous settings
- Clear affordances guiding correct use
- Fail-Safe Defaults
- Safe state on power loss
- Conservative default settings
- Automatic safety checks on startup
- Robust Margin Design
- Components rated 2x expected stress
- Algorithms stable across ±20% parameter variation
- Interfaces usable at vision/dexterity extremes
Best Practice 5: Engage Regulatory Early
When to Engage FDA
- Q-Submission for classification questions
- Pre-Submission for pathway confirmation
- Breakthrough Device designation evaluation
- Human factors protocol review
Regulatory Integration Timeline
- Phase 0: Classification research
- Phase 1: Pathway determination
- Phase 2: Standards identification
- Phase 3: Pre-Submission preparation
- Phase 4: Pre-Submission meeting
- Phase 5: Submission planning
Success Metrics for Early Development
Track these metrics to evaluate early development effectiveness:
Success Metrics Checklist
## Success Metrics for Early Development ### User Research Metrics - Users observed: Target ≥10 per user group - Use environments studied: ≥3 diverse settings - User needs identified: 40-60 typical range - Needs validation rate: >80% confirmed by multiple users ### Risk Management Metrics - Hazards identified in PHA: 20-40 typical - Risk controls per hazard: Average 2-3 - Unacceptable risks: 0 before proceeding - Risk control effectiveness: >10x risk reduction ### Design Input Metrics - Requirements per user need: 1-3 typical - Verifiable requirements: 100% - Requirements stability: <10% change after approval - Orphan requirements: 0% ### Formative Evaluation Metrics - Iterations completed: ≥3 before validation - Use errors per iteration: Decreasing trend - Task success rate: >80% by final iteration - User satisfaction: Improving trend
Appendices
Appendix A: Regulatory Resources
FDA Guidance Documents
- Design Control Guidance for Medical Device Manufacturers (March 1997)
- Link: FDA.gov/regulatory-information/search-fda-guidance-documents
- Key sections for early development: Introduction, Sections B & C
- Applying Human Factors and Usability Engineering to Medical Devices (February 2016)
- Critical for user research and formative evaluation
- Sections 5-6 most relevant for early stages
- General/Specific Intended Use - Guidance for Industry (November 1998)
- Essential for regulatory strategy
- Includes decision flowchart and examples
Recognized Standards
- ISO 14971:2019 - Risk management
- FDA Recognition #5-125
- Clauses 4-5 for early development
- IEC 62366-1:2015+AMD1:2020 - Usability engineering
- FDA Recognition #5-129
- Sections 4-5 for use specification
- ISO 13485:2016 - Quality management systems
- Section 7.3 for design controls
- Becoming QMSR requirement in 2026
Appendix B: Glossary of Terms
## Glossary of Terms **Critical Task:** A user task which, if performed incorrectly or not performed at all, would or could cause serious harm to the patient or user. **Design Controls:** Systematic methodology required by FDA for medical device development, documented in 21 CFR 820.30. **Design History File (DHF):** Compilation of records containing the design history of a finished device. **Design Input:** Physical and performance requirements of a device that are used as a basis for device design. **Design Output:** Results of the design effort at each design phase and at the end of total design effort. **Formative Evaluation:** User interface evaluation conducted during development to inform design. **Hazard:** Potential source of harm. **Hazardous Situation:** Circumstance in which people, property, or the environment are exposed to hazard(s). **Intended Use:** General purpose of the device or its function, encompassing the indications for use. **Preliminary Hazard Analysis (PHA):** Early systematic examination of potential hazards before detailed design. **Risk:** Combination of the probability of occurrence of harm and the severity of that harm. **Risk Control:** Measures implemented to reduce risk to acceptable levels. **Use Environment:** Actual conditions and settings in which users interact with the medical device. **Use Error:** User action or lack of user action while using the medical device that leads to a different result than intended. **Use Specification:** Document per IEC 62366-1 defining intended medical indication, patient population, user profile, use environment, and operating principle. **User Need:** High-level statement of what users need to accomplish, independent of design solution. **Validation:** Confirmation by examination and provision of objective evidence that the particular requirements for a specific intended use can be consistently fulfilled. **Verification:** Confirmation by examination and provision of objective evidence that specified requirements have been fulfilled.
Appendix C: Document Control Best Practices
Version Control System
## Document Version Control ### Version Numbering X.Y.Z format where: - X = Major version (significant changes) - Y = Minor version (moderate changes) - Z = Editorial version (typos, formatting) ### Version History Table | Version | Date | Author | Changes | Approver | |---------|------|--------|---------|----------| | 0.1 | [Date] | [Name] | Initial draft | N/A | | 0.2 | [Date] | [Name] | Added sections 3-4 | N/A | | 1.0 | [Date] | [Name] | First approved version | [Name] | ### Document Status Definitions - **Draft:** Under development, not for use - **For Review:** Complete, awaiting approval - **Approved:** Released for use - **Obsolete:** Superseded, retained for history
Appendix D: Early Development Checklist
## Early Development Phase Checklist ### Phase 0 Complete - [ ] Market need validated with >10 potential users - [ ] Technical feasibility demonstrated - [ ] Competitive analysis completed - [ ] Go/No-go decision documented - [ ] Development team identified - [ ] Funding secured ### Phase 1 Complete - [ ] Intended use statement drafted - [ ] Indications for use defined - [ ] User profiles documented - [ ] Use environments characterized - [ ] Use specification per IEC 62366-1 ### Phase 2 Complete - [ ] User needs documented and prioritized - [ ] Preliminary hazard analysis completed - [ ] Known problems research done - [ ] Initial risk controls identified ### Phase 3 Complete - [ ] Design and development plan approved - [ ] Design inputs documented - [ ] Requirements traceable to needs - [ ] Verification methods identified - [ ] Design input review completed - [ ] Change control initiated ### Phase 4 Complete - [ ] Formative evaluations conducted (≥3 iterations) - [ ] Critical tasks validated - [ ] Risk controls defined - [ ] Design iterations documented ### Phase 5 Complete - [ ] Traceability matrix established - [ ] HFE file organized - [ ] DHF structure created - [ ] Documentation control system operational - [ ] Ready for detailed design phase
Conclusion
This guide provides a comprehensive framework for navigating the critical early stages of medical device development. By following these structured approaches to user research, requirements development, and risk management, startups can build a strong foundation for successful product development while maintaining regulatory compliance.
Remember that early-stage development is inherently iterative. The templates and processes provided here should be adapted to your specific device, users, and regulatory pathway. The key is to maintain discipline in documentation and decision-making while remaining flexible enough to respond to new insights from user research and formative evaluations.
Success in medical device development requires balancing innovation with safety, user needs with technical constraints, and speed to market with regulatory requirements. This guide provides the tools to achieve that balance, but ultimately success depends on committed execution and continuous learning from users, regulators, and the market.
As you progress beyond these early stages into detailed design, verification, and validation, maintain the user focus and risk-based thinking established here. The foundation you build in these early phases will determine not just regulatory success, but ultimately whether your device truly improves patient care and user experience.
This guide represents current FDA guidance and recognized standards as of the publication date. Always verify current regulatory requirements and consult with regulatory professionals for device-specific guidance.
Document Version: 1.0
Last Updated: 2024
Total Word Count: ~18,000 words
Need help implementing these strategies for your device?
Our medical device experts can guide you through every step of regulatory compliance and development.